Welcome to the Galaxy Genome Lab. Get quick access to tools, workflows and tutorials for genome assembly and annotation.
What is this page?
Galaxy
This site genome.usegalaxy.org is connected to the same server as usegalaxy.org, but with an interface dedicated to helping our researchers. Your history, jobs and data quota are shared with the "Base" website.

Data import and preparation
If you are new to galaxy, uploading your data is a good place to start!
Check out the Tools and Workflows tabs for different approaches to uploading data.
You can upload your data to Galaxy using the Upload tool from anywhere in Galaxy. Just look for the "Upload data" button at the top of the tool panel.
We recommend subsampling large data sets to test tools and workflows. A useful tool is seqtk_seq, setting the parameter at "Sample fraction of sequences".
No, do not upload personal or sensitive, such as human health or clinical data. Please see our Data Privacy page for definitions of sensitive and health-related information.
Please also make sure you have read our Terms of Service, which covers hosting and analysis of research data.
Please read our Privacy Policy for information on your personal data and any data that you upload.
Quality control and data cleaning is an essential first step in any NGS analysis. This tutorial will show you how to use and interpret results from FastQC, NanoPlot and PycoQC.
This practical aims to familiarize you with the Galaxy user interface. It will teach you how to perform basic tasks such as importing data, running tools, working with histories, creating workflows, and sharing your work.
Any user of Galaxy can request support through an online form.
Common tools are listed here, or search for more in the full tool panel to the left.
A workflow is a series of Galaxy tools that have been linked together to perform a specific analysis. You can use and customize the example workflows below. Learn more.
Genome assembly
Common tools are listed here, or search for more in the full tool panel to the left.
YAHS is a scaffolding tool based on a computational method that exploits the genomic proximity information in Hi-C data sets for long-range scaffolding of de novo genome assemblies. Inputs are the primary assembly (or haplotype 1), and HiC reads mapped to the assembly. See this tutorial to learn how to create a suitable BAM file.
Input data:
fasta
|
Primary assembly or Haplotype 1 genome.fasta |
bam
|
HiC reads mapped to assembly mapped_reads.bam |
BUSCO: assessing genome assembly and annotation completeness with Benchmarking Universal Single-Copy Orthologs. The tool attempts to provide a quantitative assessment of the completeness in terms of the expected gene content of a genome assembly, transcriptome, or annotated gene set.
Input data:
fasta
|
A workflow is a series of Galaxy tools that have been linked together to perform a specific analysis. You can use and customize the example workflows below. Learn more.
TSI assembly workflows - PacBio HiFi or Nanopore data
This How-to-Guide describes the steps required to assemble your genome on Galaxy, using multiple workflows. There is also a guide about the Genome Assessment workflow, and the HiC Scaffolding workflow.
General assembly workflows - Nanopore and Illumina data
This tutorial describes the steps required to assemble a genome on Galaxy with Nanopore and Illumina data.
VGP assembly workflows - PacBio HiFi and (optional) HiC data
These workflows have been developed as part of the global Vertebrate Genome Project (VGP). A guide to using these in Galaxy can be found here. A complete guide to the individual workflows and sample results can be found here. There are many different ways that these workflows can be used in practice - for a comprehensive example, check out this Galaxy tutorial.
This workflow produces a Meryl database and Genomescope outputs that will be used to determine parameters for following workflows, and assess the quality of genome assemblies. Specifically, it provides information about the genomic complexity, such as the genome size and levels of heterozygosity and repeat content, as well about the data quality.
Recommended: v0.1.7 (recently tested).
Input data:
fastq
|
PacBio HiFi reads |
This workflow uses hifiasm (HiC mode) to generate HiC-phased haplotypes (hap1 and hap2). This is in contrast to its default mode, which generates primary and alternate pseudohaplotype assemblies. This workflow includes three tools for evaluating assembly quality: gfastats, BUSCO and Merqury.
Note: if you have multiple input files for each HiC set, they need to be concatenated. The forward set needs to be concatenated in the same order as reverse set.
Recommended: v0.1.10 (recently tested).
Input data:
fasta
|
PacBio HiFi reads |
fastq
|
PacBio HiC reads (forward) |
fastq
|
PacBio HiC reads (reverse) |
meryldb
|
Meryl kmer database |
txt
|
GenomeScope genome profile summary |
This workflow uses hifiasm to generate primary and alternate pseudohaplotype assemblies. This workflow includes three tools for evaluating assembly quality: gfastats, BUSCO and Merqury.
Recommended: v0.3.5 (recently tested).
Input data:
fasta
|
PacBio HiFi reads |
meryldb
|
Meryl kmer database |
txt
|
GenomeScope genome profile summary |
Yes. Galaxy has assembly tools for small prokaryote genomes as well as larger eukaryote genomes. We are continually adding new tools and optimising them for large genome assemblies - this means adding enough computer processing power to run data-intensive tools, as well as configuring aspects such as parallelisation.
Please contact us if:
- there is a tool you wish to request
- a tool appears to be broken or running slowly
- See the tutorials in this Help section. They cover different approaches to genome assembly.
- Read the methods in scientific papers about genome assembly, particularly those about genomes with similar characteristics to those in your project
- See the Workflows section for examples of different approaches to genome assembly - these cover different sequencing data types, and a variety of tools.
Genome assembly can be a very involved process. A typical genome assembly procedure might look like:
- Data QC - check the quality and characteristics of your sequencing reads.
- Kmer counting - to determine genome characteristics such as ploidy and size.
- Data preparation - trimming and filtering sequencing reads if required.
- Assembly - for large genomes, this is usually done with long sequencing reads from PacBio or Nanopore.
- Polishing - the assembly may be polished (corrected) with long and/or short (Illumina) reads.
- Scaffolding - the assembly contigs may be joined together with other sequencing data such as HiC.
- Assessment - at any stage, the assembly can be assessed for number of contigs, number of base pairs, whether expected genes are present, and many other metrics.
- Annotation - identify features on the genome assembly such as gene names and locations.
A graphical representation of genome assembly
There is no best set of tools to recommend - new tools are developed constantly, sequencing technology improves rapidly, and many genomes have never been sequenced before and thus their characteristics and quirks are unknown. The "Tools" tab in this section includes a list of commonly-used tools that could be a good starting point. You will find other tools in recent publications or used in workflows.
You can also search for tools in Galaxy's tool panel.
We recommend testing a tool on a small data set first and seeing if the results make sense, before running on your full data set.
Once a genome has been assembled, it is important to assess the quality of the assembly, and in the first instance, this quality control (QC) can be achieved using the workflow described here.
Any user of Galaxy can request support through an online form.
Genome annotation
Common tools are listed here, or search for more in the full tool panel to the left.
MAKER is able to annotate both prokaryotes and eukaryotes. It works by aligning as many evidences as possible along the genome sequence, and then reconciling all these signals to determine probable gene structures.
The evidences can be transcript or protein sequences from the same (or closely related) organism. These sequences can come from public databases (like NR or GenBank) or from your own experimental data (transcriptome assembly from an RNASeq experiment for example). MAKER is also able to take into account repeated elements.
Input data:
fasta
|
Genome assembly |
fasta
|
Protein evidence (optional) |
RepeatMasker is a program that screens DNA for repeated elements such as tandem repeats, transposons, SINEs and LINEs. Galaxy should have the full and curated DFam screening databases, or a custom database can be provided in fasta format. Additional reference data can be downloaded from RepBase.
Input data:
fasta
|
Genome assembly |
A workflow is a series of Galaxy tools that have been linked together to perform a specific analysis. You can use and customize the example workflows below. Learn more.
General use
Annotates a genome using multiple rounds of Maker, including gene prediction using SNAP and Augustus. Based on the GTN Maker tutorial, with an updated version of BUSCO.
Tools: maker snap augustus busco jbrowse
Input data:
fasta
|
Genome assembly |
fasta
|
EST and/or cDNA sequences |
fasta
|
Protein sequences |
Transcript alignment
This How-to-Guide will describe the steps required to align transcript data to your genome on Galaxy, using multiple workflows.
These slides from the Galaxy training network explain the process of genome annotation in detail. You can use the ← and → keys to navigate through the slides.
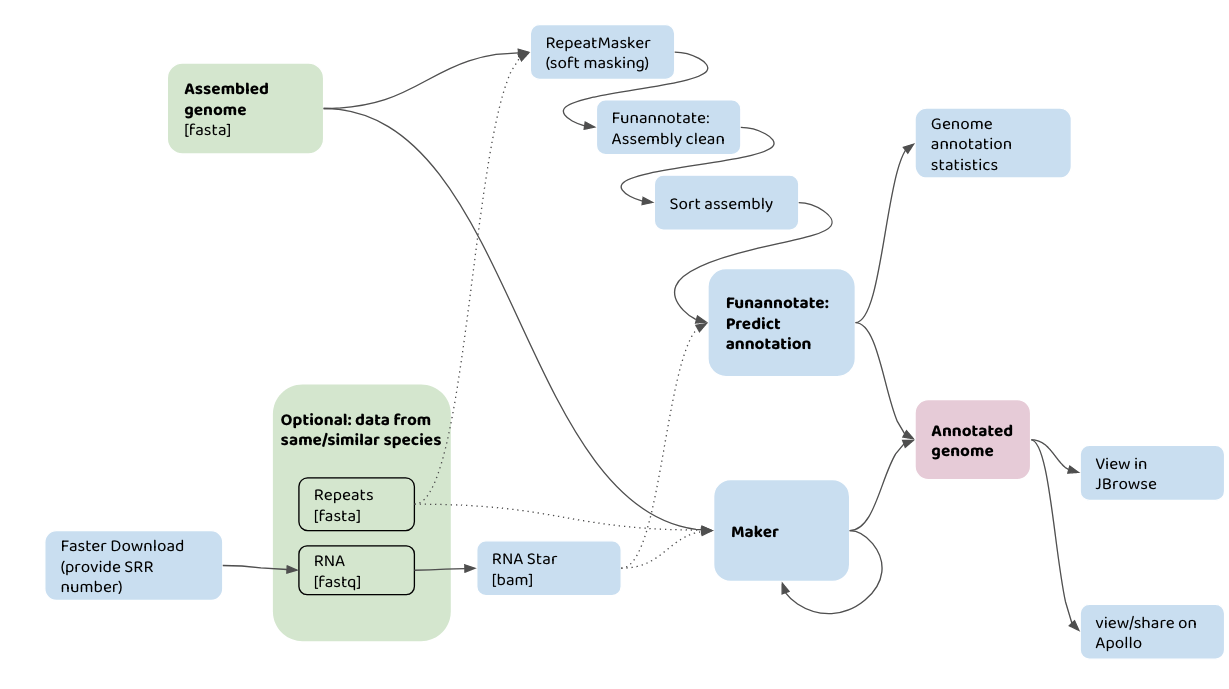
The flowchart below shows how you might use your input data (in green) with different Galaxy tools (in blue) to annotate a genome assembly. For example, one pathway would be taking an assembled genome, plus information about repeats, and data from RNA-seq, to run in the Maker pipeline. The annotatations can then be viewed in JBrowse.
A graphical representation of genome annotation
Genome annotation with Maker
Genome annotation of eukaryotes is a little more complicated than for prokaryotes: eukaryotic genomes are usually larger than prokaryotes, with more genes. The sequences determining the beginning and the end of a gene are generally less conserved than the prokaryotic ones. Many genes also contain introns, and the limits of these introns (acceptor and donor sites) are not highly conserved. This Galaxy tutorial uses MAKER to annotate the genome of a small eukaryote: Schizosaccharomyces pombe (a yeast).
Any user of Galaxy can request support through an online form.
Contributors

