Galaxy Labs YAML schema
This document describes the structure and validation rules for a
Galaxy Lab section.yml file.
A minimal example of the full schema is shown below, but a real example
would have more tabs and content items. Typically there should be only
one section per YAML file (e.g.
section_1.yml,
section_2.yml).
id: section_1
title: Example section
tabs:
- id: tools
title: Tools
heading_md: Common tools are listed here, or search for more in the full tool panel to the left.
content:
- buttons:
- icon: run
link: "{{ galaxy_base_url }}/tool_runner?tool_id=toolshed.g2.bx.psu.edu/repos/devteam/fastqc/fastqc"
title_md: `FastQC` - sequence quality reports
description_md: |
Before using your sequencing data, it’s important to ensure that
the data quality is sufficient for your analysis.
inputs:
- label: Sequencing data for analysis
datatypes:
- fasta
- fastq
- bam
- sam
- id: workflows
title: Workflows
heading_md: |
A workflow is a series of Galaxy tools that have been linked together
to perform a specific analysis. You can use and customize the example workflows
below.
<a href="https://galaxyproject.org/learn/advanced-workflow/">
Learn more
</a>.
content:
- title_md: Data QC
description_md: |
Report statistics from sequencing reads.
**Tools**
- `nanoplot`
- `fastqc`
- `multiqc`
buttons:
- icon: run
link: "{{ galaxy_base_url }}/workflows/trs_import?trs_server=workflowhub.eu&run_form=true&trs_id=222"
tip: Import to Galaxy AU
- icon: view
link: https://workflowhub.eu/workflows/222
tip: View in WorkflowHub
The above YAML results in the following element being rendered:
Example section
A workflow is a series of Galaxy tools that have been linked together to perform a specific analysis. You can use and customize the example workflows below. Learn more .
You can upload your data to Galaxy using the Upload tool from anywhere in Galaxy. Just look for the "Upload data" button at the top of the tool panel.
We recommend subsampling large data sets to test tools and workflows.
A useful tool is
seqtk_seq,
setting the parameter at "Sample fraction of sequences"
The YAML data above can be broken down into the following components:
The YAML schema expects each of items 1-6 to be a list of items, so these elements can be repeated to provide as many Tabs, Content Items etc. as you like.
Section
Description
A Galaxy Lab section - this is the root of the section.yml
file.
Properties
-
id(string, required)
Unique identifier for the section. -
title(string, required)
Title of the section. -
tabs(array of SectionTab, required)
List of tabs to be displayed for this section. -
exclude_from(array of strings or null, optional)
List of Galaxy domain names (e.g.usegalaxy.eu) that this section should be excluded from e.g. if a feature is not implemented or tools are not available. Defaults to an empty array.
Example
id: section_1
title: Example section
tabs:
- <SectionTab>
- <SectionTab>
Section Tab
Description
A tab with a list of expandable items to be shown in the section.
Properties
-
id(string, required)
Unique identifier for the tab. -
title(string or null, optional)
Title of the tab. -
content(array of Content Items, object, or null, optional)
A list of content items to be displayed in this tab. This can be omitted to create a tab with arbitrary MD/HTML content using theheading_mdproperty. Can be any of:- An array of Content Items.
- An object of { 'subsections': array of TabSubsections. }
null.
-
heading_md(string or null, optional)
Markdown or HTML formatted string for the heading. The value can be any length, so can even be extended to create a block of MD/HTML content. -
exclude_from(array of strings or null, optional)
List of Galaxy domain names (e.g.usegalaxy.eu) that this tab should be excluded from e.g. if a feature is not implemented or tools are not available. Defaults to an empty array.
Example
id: section_1_tools
title: Tools
heading_md: |
These tools will get you started with your analysis.
More advanced tools are available in the
[next section](#advancedSection).
content:
- <ContentItem>
- <ContentItem>
id: section_1_tools
title: Tools with subsections
heading_md: |
These tools are listed in subsections for easier navigation.
content:
subsections:
- <TabSubsection>
- <TabSubsection>
Tab Subsection
Description
A content subsection, to be used for dividing content into subsections, each with a title. e.g. "Short-read", "Long-read", "Hybrid".
Properties
-
id(string, required)
Unique identifier for the subsection. -
title(string, required)
Title of the subsection. -
content(array of ContentItem, required)
Content of the subsection. -
exclude_from(array of strings or null, optional)
List of Galaxy domain names (e.g.usegalaxy.eu) that this tab subsection should be excluded from e.g. if a feature is not implemented or tools are not available. Defaults to an empty array.
Example
id: tools_short_read
title: These tools are optimized for short-read analysis
content:
- <ContentItem>
- <ContentItem>
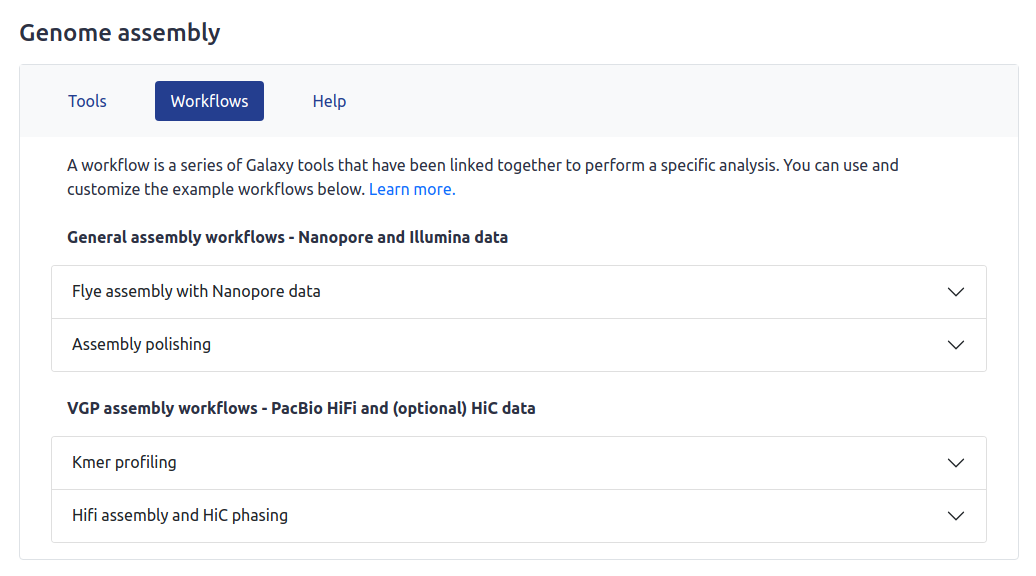
An example of this can be found on the Genome Lab, where subsections are used to separate Nanopore, Illumina and PacBio tools:
Content Item
Description
A content item, which is typically a Galaxy tool, workflow or tutorial.
In the UI, this will be rendered as an "accordion" item. If buttons are not
specified, no buttons will be displayed. For tools and
workflows, inputs can be used to describe the essential inputs.
When inserting URLs for the Galaxy server (e.g. link to Galaxy tool),
ensure that you make use of the {{ galaxy_base_url }}
variable to ensure that the link is correctly generated for different
Galaxy servers! See below for an example.
Properties
-
title_md(string, required)
Markdown or HTML formatted title. -
description_md(string, required)
Markdown or HTML formatted description. -
button_link(deprecated in favour ofbuttons) (string or null, optional)
URL for the button link - this is typically used as a "run" button for tools or workflows, but can be customized by setting the otherbutton_*properties. -
button_tip(deprecated in favour ofbuttons) (string or null, optional)
Tooltip text for the button - this will be shown when the user's mouse passes over the item. -
button_md(deprecated in favour ofbuttons) (string or null, optional)
Markdown or HTML formatted string to display inside the "button" button. -
button_icon(deprecated in favour ofbuttons) (ButtonIcon or null, optional)
Icon for the button - must be one of the accepted ButtonIcon strings. -
view_link(deprecated in favour ofbuttons) (string or null, optional)
URL for the "view" link - this is typically a link to view more information about the item e.g. a workflow in WorkFlowHub. -
view_tip(deprecated in favour ofbuttons) (string or null, optional)
Tooltip text for the view - this will be shown when the user's mouse passes over the item. -
view_md(deprecated in favour ofbuttons) (string or null, optional)
Markdown or HTML formatted string to display inside the "view" button. -
view_icon(deprecated in favour ofbuttons) (ButtonIcon or null, optional)
Icon for the "view" button - must be one of the accepted ButtonIcon strings. -
buttons(array of Button items or null, optional)
A list of buttons to display for this item. -
exclude_from(array of strings or null, optional)
List of Galaxy domain names (e.g.usegalaxy.eu) that this item should be excluded from e.g. if a feature is not implemented or a tool is not available. Defaults to an empty array.
Example
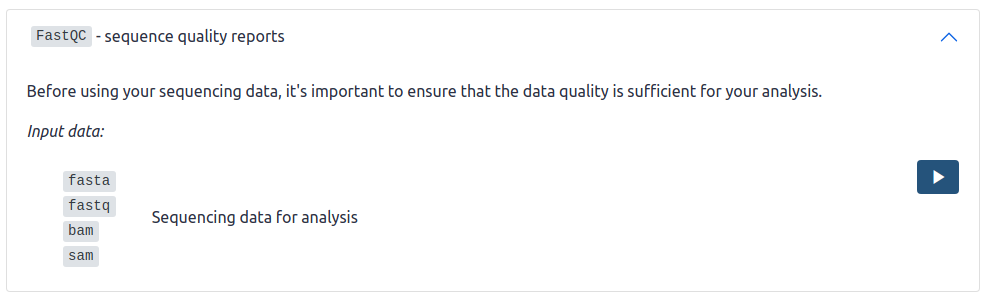
- title_md: `FastQC` - sequence quality reports
description_md: |
Before using your sequencing data, it’s important to ensure that
the data quality is sufficient for your analysis.
buttons:
- icon: run
link: "{{ galaxy_base_url }}/tool_runner?tool_id=toolshed.g2.bx.psu.edu/repos/devteam/fastqc/fastqc"
inputs:
- <ItemInput>
- <ItemInput>
The above YAML is rendered as:
- title_md: Data QC on Galaxy AU
description_md: |
Report statistics from sequencing reads.
**Tools**
- `nanoplot`
- `fastqc`
- `multiqc`
buttons:
- icon: run
link: "{{ galaxy_base_url }}/workflows/trs_import?trs_server=workflowhub.eu&run_form=true&trs_id=222"
tip: Import to Galaxy Australia
- icon: view
link: https://workflowhub.eu/workflows/222
tip: View in WorkflowHub
exclude_from:
- usegalaxy.org
- usegalaxy.eu
The above YAML is rendered as:
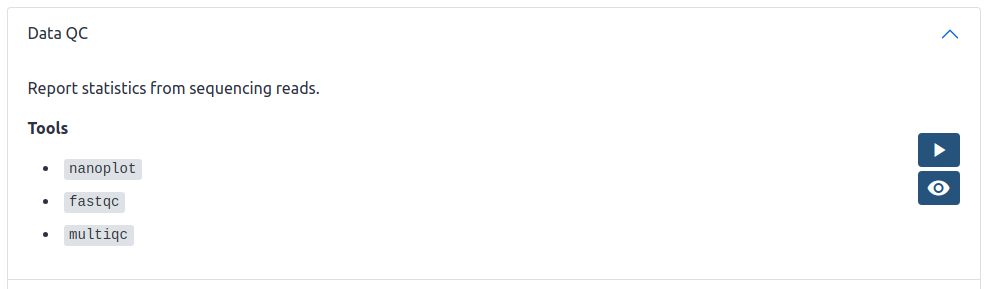
You can create a workflow import URL as shown above from either DockStore or Workflowhub by changing the TRS ID and TRS server. The TRS ID can be found at the end of the URL of the workflow page on DockStore or Workflowhub.
InputItem
Description
An input dataset, typically for a Galaxy tool or workflow. The list of inputs will be displayed as a table.
Properties
-
datatypes(array of strings, optional)
List of accepted Galaxy datatypes for this input dataset. e.g.['fastqsanger', 'fasta']. -
label(string ornull, optional)
Short description for the input dataset e.g. "Raw reads".
Example
label: Raw reads
datatypes:
- fasta
- fastqsanger
Button
Description
A button to be displayed with a link, containing either an icon or markdown. Can optionally have a tooltip that will be shown when the user's mouse passes over the button.
Properties
-
link(string, required)
The URL that the button will navigate to when clicked. -
label_md(string or null, optional)
Markdown or HTML formatted label to display inside the button. If omitted, only the icon will be shown. -
icon(ButtonIcon or null, optional)
Icon to display in the button. Must be one of the accepted ButtonIcon values. If provided, this will be shown instead of the label. -
tip(string or null, optional)
Tooltip text to display when the user hovers over the button.
ButtonIcon
Description
Accepted icons identifiers for buttons.
run |
|
tutorial |
|
social |
|
help |
|
view |